





Abstract
Il microbiota del tratto gastrointestinale (GIT) dei ruminanti ha un ruolo fondamentale sulla salute dell’animale e sulla sua efficienza produttiva. L’idea di poter intervenire modificando la struttura e la composizione del microbiota, tramite la somministrazione di diete diverse, nasce dalla consapevolezza che la composizione delle comunità microbiche presenti in questo tratto è sensibile a fattori endogeni ed esogeni. Inoltre, poter modellare il microbiota del GIT potrebbe avere importanti ripercussioni non solo sulla salute e l’efficienza produttiva dell’animale ma anche sull’emissioni di metano (CH4). Questo studio, quindi, si pone molteplici obiettivi; in primo luogo quello di identificare e caratterizzare il microbiota del GIT bufalino, ed in seguito quello di verificare se la somministrazione di due diete che differiscono per il rapporto CNS (carboidrati non strutturali)/PG (proteine grezze) influenzi o meno la struttura della comunità microbica del rumine, intestino e feci.
Introduzione
L’interazione simbiotica tra i mammiferi ed i loro diversi microrganismi è di notevole importanza in quanto tale mutualismo fornisce nutrimento, sviluppo e benefici per il sistema immunitario dell’ospite (1). In particolare, nei ruminanti, il tratto gastrointestinale (GIT) ospita numerose e diverse comunità batteriche che assumono un ruolo fondamentale nella degradazione di proteine, grassi, amido, cellulosa, emicellulosa e lignina (2). Di notevole importanza è il ruolo dei microrganismi presenti nel rumine e nell’intestino in quanto provvedono alle fermentazioni di polisaccaridi complessi attraverso la secrezione di specifici enzimi che l’animale non riesce a sintetizzare autonomamente. I prodotti della degradazione microbica sono gli acidi grassi volatili a catena corta (VFAs) come l’acetato, il butirrato e il propionato che fungono da principale fonte di energia per i ruminanti. Attraverso le reazioni di fermentazione, a livello ruminale, vengono prodotti anche anidride carbonica (CO2) e metano (CH4), comportando sia emissioni di gas serra sia una perdita dell’energia totale proveniente dalla fermentazione del cibo. Il microbiota del GIT, inoltre, influenza fortemente lo sviluppo e la maturazione del sistema immunitario dell’ospite e può modulare le risposte immunitarie sia innate che adattative.
La composizione del microbioma del GIT può essere estremamente variabile e comprende per lo più batteri ma sono presenti anche archei, protozoi, funghi e virus (3). La sua composizione, intesa come abbondanza o meno di diverse comunità di microrganismi, può variare a seconda di fattori legati sia a parametri intrinseci dell’ospite, come il pH ruminale, lo stato nutrizionale e l’età dell’animale, sia in base alle condizioni ambientali in cui l’ospite vive (1). Recenti studi hanno dimostrato, inoltre, che la composizione del microbiota può variare anche in base alla frequenza con la quale vengono alimentati gli animali e alla capacità di ingestione del cibo (4). Tali evidenze suggeriscono la possibilità di modellare il microbiota del GIT in base alla dieta somministrata all’animale.
Poter favorire determinate comunità microbiche rispetto ad altre potrebbe promuovere l’efficienza produttiva dell’animale (5-7) e ridurre le emissioni di gas serra (8,9). Tali obiettivi sono in linea con quelli proposti a livello globale. Ad oggi, infatti, la crescente domanda di alimenti dovuta all’incremento della popolazione mondiale può essere soddisfatta esclusivamente attraverso sistemi produttivi altamente efficienti e sostenibili dal punto di vista ambientale ed economico. La resa produttiva dei ruminanti è in linea generale minore rispetto a quella dei monogastrici; per tale motivo scegliere di alimentare questi animali con i sottoprodotti dell’industria alimentare potrebbe incrementare la sostenibilità del sistema produttivo. I residui dell’industria molitoria, delle distillerie e delle birrerie sono alimenti già utilizzati nelle diete dei ruminanti (10). Tali sottoprodotti possono essere facilmente conservati attraverso tecniche di insilamento ed essiccazione così da essere disponibili anche su periodi più lunghi dell’anno ed aggiunti in seguito alle razioni alimentari. I processi di conservazione, inoltre, intervengono anche sulla composizione chimica dell’alimento, da un lato migliorandone il contenuto proteico, dall’altro potenzialmente promuovendo lo sviluppo di fattori anti-nutrizionali, micotossine e diossine (11). Studi recenti sull’utilizzazione di scarti dell’industria molitoria e buccette di pomodoro nell’alimentazione delle capre, hanno riportato dei buoni risultati per quanto riguarda la produzione di latte, la sua composizione e le emissioni di metano, senza intaccare la digeribilità e il livello di ingestione della dieta (12).
Le buccette di pomodoro, nel Sud Italia, rappresentano un sottoprodotto particolarmente abbondante data la grossa quantità di aziende che trasformano questo alimento in polpa. Studi sull’utilizzo di questo prodotto nella dieta dei bufali sono stati già condotti, confermandolo come valida alternativa all’alimentazione tradizionale (12). È stato anche dimostrato che inserendo le buccette nella dieta si riducono le emissioni di metano (12). L’avvento delle tecnologie di sequenziamento ad alto rendimento, inoltre, ha reso possibile la caratterizzazione di specie batteriche presenti in diverse matrici, consentendo così l’identificazione e la classificazione di tutte le specie presenti, comprese quelle dei microbi non coltivabili (13). Questo approccio si è rivelato estremamente adatto allo studio del microbiota ruminale del GIT poiché è stato stimato che solo il 20% di tali comunità complesse possono essere coltivate con pratiche standard (14). Gli studi basati sul sequenziamento del gene 16S rRNA consentono l’identificazione di specie poco abbondanti all’interno della popolazione. Quindi questa tecnica rende possibile la caratterizzazione dei cambiamenti nelle comunità microbiche complesse in risposta a fattori esterni, come la dieta.
Pertanto, gli obiettivi di questo studio sono quelli di caratterizzare il differente microbiota del GIT nelle bufale in diversi siti corporei (rumine, intestino e feci) e valutare l’influenza sul microbiota nel tratto gastrointestinale delle bufale di una razione che presenta un differente rapporto carboidrati non strutturali (NSC)/proteine grezze (PG), rispetto a quella tradizionale, in seguito all’aggiunta di buccette di pomodoro insilate.
Materiali e metodi
L’esperimento è stato condotto su 20 bufale in asciutta, con un’età media di 7.1± 1.2 anni e ordine di parto di 3.7 ± 1.3 anni, per le quali era già prevista la macellazione. Gli animali sono stati allevati in paddock con pavimento pieno, divisi in due gruppi omogenei e alimentati con razioni hanno caratterizzate da diverso rapporto carboidrati non strutturali e proteine grezze (CNS/PG). Nella razione tradizionale, il rapporto era di 1.9, mentre in quella alternativa di 2.3. Per entrambi i gruppi la dieta è stata somministrata per 7 settimane (3 di adattamento e 4 di sperimentazione). Per valutare la digeribilità della dieta sono stati raccolti dei campioni di feci per ogni animale per 5 giorni consecutivi. Alla fine del periodo di sperimentazione le bufale sono state macellate e sono stati raccolti 20 campioni di rumine, intestino e feci per le analisi molecolari. Al fine di individuare il microbiota dei siti d’interesse sono stati eseguiti l’amplificazione ed il sequenziamento del gene 16S rRNA mediante PCR. I risultati ottenuti sono stati analizzati e processati tramite software ad hoc per la classificazione tassonomica. Il secondo step è stato quello di valutare le caratteristiche del microbiota attraverso analisi afferenti alla statistica delle popolazioni utilizzando l’”alpha diversity analysis” e la “beta diversity analysis“. In ultimo è stata eseguita un’analisi ANCOM (analisi della composizione dei microbiomi).
Risultati e discussione
L’analisi tassonomica dei microrganismi presenti nel rumine, intestino e feci degli animali alimentati in maniera tradizionale ed alternativa indica che, in tutti i campioni di entrambi i gruppi, i tre phyla più abbondanti sono i Bacteroides, Firmicutes e Proteobacteria, con diversa abbondanza relativa tra i distretti corporei esaminati, come è possibile osservare nella figura 1.
Figura 1. Abbondanza relativa (frequenza relativa media) dei Phyla batterici identificati in diversi siti GIT (rumine, intestino e feci) delle bufale alimentate in maniera tradizionale (n = 10) e alternativa (n = 10).
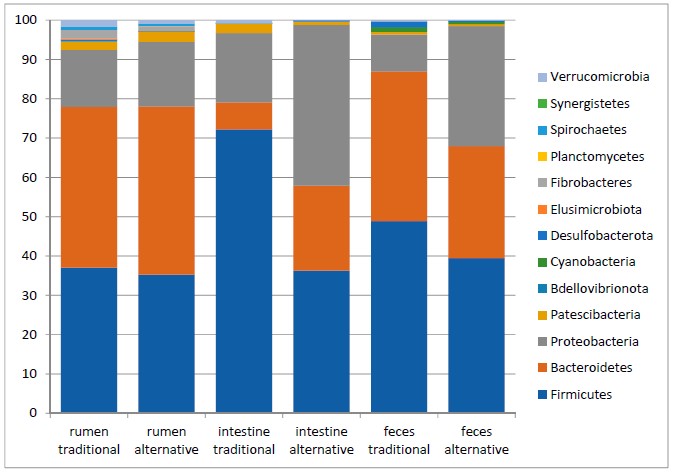
Il phylum dei Bacteroides è molto abbondante nel rumine ed include principalmente Prevotellacae e Rikinellacae. Questi microrganismi sono in grado di degradare gli oligominerali, i polisaccaridi e altri substrati come la pectina e la mucina (8,15–19). Il phylum Firmicutes è più abbondante nell’intestino e nelle feci. Le famiglie più rappresentative sono rispettivamente Peptostreptococcaceae, Erysipelotrichaceae, Christensenellaceae, Clostridiaceae e Lachnospiraceaee Oscillospiraceae, mentre nelle feci questo Phylum include per la maggior parte Lachnospiraceae, Oscillospiraceae, UCG-010, Christensenellaceae ed il Coprostanoligenes_group. Il compito principale di questi batteri è quello di degradare la componente fibrosa della razione. Questi due Phyla sono composti principalmente da batteri Gram-negativi che sono in grado di deteriorare la parete cellulare delle piante secernendo enzimi come il FAE (Ferulolil esterasi) che migliorano la digeribilità di diete molto fibrose (2).
Nel rumine, invece, i risultati hanno evidenziato la presenza di un microbiota composto principalmente da taxa come Prevotella, Bacteroidales UCG-001, Christensenellaceae, Rikenellaceae RC9_gut_group, Ruminococcaceae NK4A214_group, Oscillospiraceae, Ruminococcused Eubatteri. Nel presente studio, questi taxa sono stati trovati anche nel microbiota caratteristico dell’intestino e delle feci, sottolineando l’importanza di queste popolazioni come membri fondamentali del microbiota del GIT della bufala. Ad ogni modo, il rumine, l’intestino e le feci hanno mostrato una composizione microbica diversa. Ad esempio, in relazione ai generi maggiormente presenti è stato visto che Ruminobacter, Butyrivibrio, Succiniclasticum, Fibrobacter, Succinivibrionaceae UCG-002, Papillibacter, gruppo intestinale Bacteroidales BS11, Succinivibrio, Saccharofermentans e Lachnospiraceae, il gruppo UCG-008 e NK3A20 sono tra quelli identificati come fortemente legati al microbiota ruminale. Nell’intestino e nelle feci, invece, sono presenti per lo più i generi Bacteroides, Alistips, Rikenellaceae, Turicibacter, Clostridium ED Escherichia-Shigella.
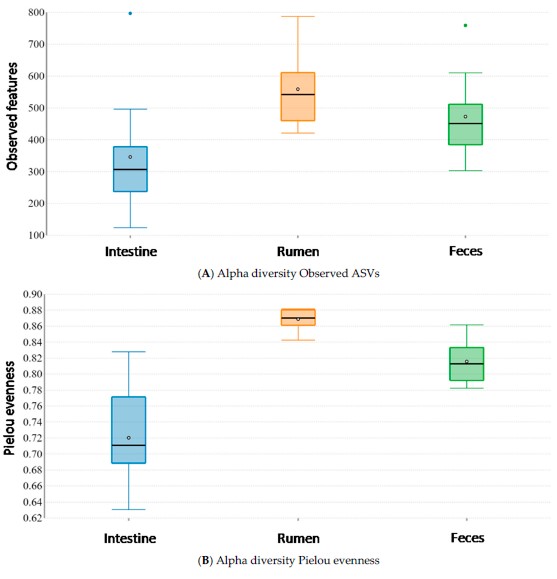
L’analisi “alpha diversity” è stata eseguita per verificare le caratteristiche della popolazione microbica all’interno di ogni sito corporeo (rumine, intestino e feci) del gruppo alimentato tradizionalmente, prendendo in considerazione due parametri: Observed ASVs, che rappresenta una misura quantitativa del numero di taxa osservati (Assigned Sequence Variants – ASVs) e l’indice Pielou eveness, che misura quanto uniformemente i taxa si ripartiscano all’interno della popolazione microbica osservata. Tali indici hanno evidenziato significative differenze tra i siti corporei sia nel numero dei diversi taxa osservati (Observed ASVs) sia per quanto riguarda l’omogeneità della popolazione (Pielou eveness). In particolare, nell’intestino è stata riscontrata una bassa uniformità della popolazione, mentre nel rumine la popolazione è risultata più omogenea (Figura 2).
Figura 2. Analisi “Alpha diversity” del microbiota di rumine, intestino e feci delle bufale alimentate con una dieta tradizionale (n = 10). Grafico A: ASVs osservati; Grafico B: Indice di Pielou eveness.
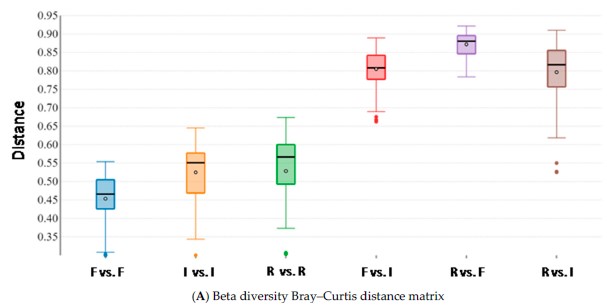
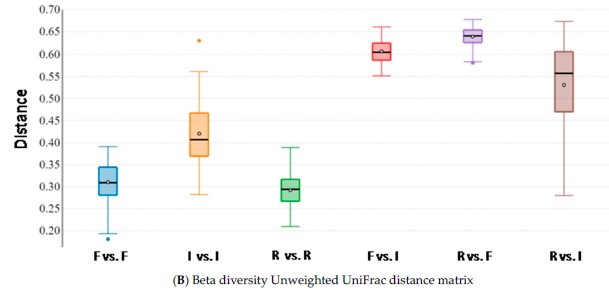
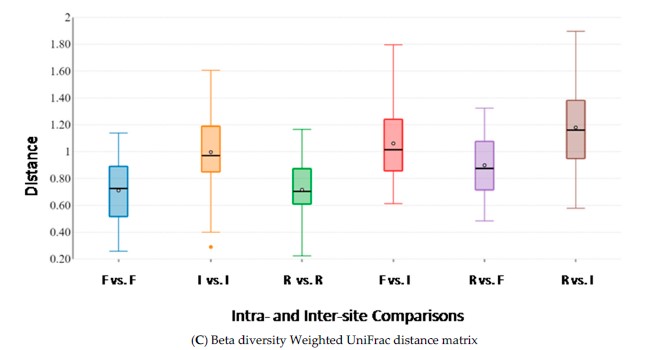
Successivamente si è passati alla valutazione della “beta analysis”, così da verificare la diversità della struttura della popolazione microbica tra i vari siti corporei ed i gruppi a diverso regime alimentare. In primis è stata valutata la dissimilarità tra i campioni dei siti appartenenti allo stesso gruppo (bufale alimentate in maniera tradizionale) utilizzando diverse matrici: Bray-Curtis, weighted e unweighted UniFrac (Figura 3). Tali indici vanno da 0 a 1, dove 0 indica che le popolazioni condividono tutte le caratteristiche, mentre 1 indica che non ne hanno nessuna in comune. Dai risultati si può notare che i campioni sono caratterizzati per lo più da bassi indici di dissimilarità; tuttavia, sono state registrate significative differenze tra i tre diversi test effettuati. Questo trend mostra che la struttura del microbiota è molto diversa tra i vari siti corporei sia in termini di taxa presenti, che nell’abbondanza o meno di diverse specie.
Figura 3. Box plot della “Beta diversity” all’interno e tra i siti corporei di bufale alimentate con la razione tradizionale (n = 10). Distanze calcolate con diverse matrici: (A) Bray–Curtis, (B) unweighted UniFrac e (C) weighted UniFrac. La lettera F indica feci, I indico intestino, R indica rumine.
Successivamente, per comparare la distanza in termini di diversità tra le comunità che compongono il microbiota tra i due gruppi (bufale alimentate in maniera tradizionale e alternativa) per ogni sito corporeo, sono state utilizzate le analisi PCOA (analisi delle coordinate principali), così da comprendere la variazione della struttura del microbiota dei vari siti tra i due gruppi a partire dalle analisi di dissimilarità svolte precedentemente (Figura 4).
Figura 4. Analisi delle coordinate (PCoA) tra bufale alimentate con la razione tradizionale (rosso) e con la razione alternativa (blu). A: rumine, B: intestino, C: feci.
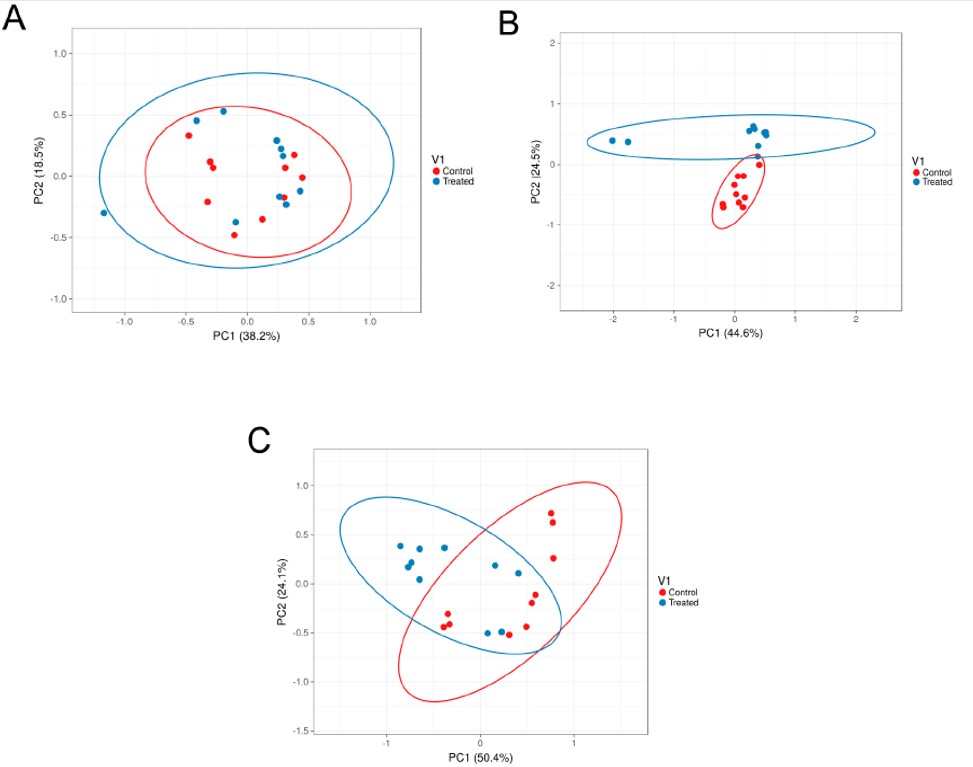
I risultati ottenuti mostrano che la composizione del microbiota del rumine dei due gruppi a diverso regime alimentare è molto simile (i campioni infatti formano un unico cluster), mentre i microbiota di intestino e feci appaiono separati (formano due cluster diversi), indicando differenze significative nella struttura della popolazione in relazione alla dieta. Infine, per ogni sito corporeo, è stata condotta un’analisi di tipo ANCOM per valutare i taxa con abbondanze significativamente diverse in funzione del regime alimentare.
Figura 5. Risultati analisi ANCOM tra bufale alimentate con una razione tradizionale (n = 10) e una razione alternativa (n = 10). I punti rappresentano le caratteristiche identificate dall’analisi ANCOM come differenzialmente abbondanti tra i gruppi.
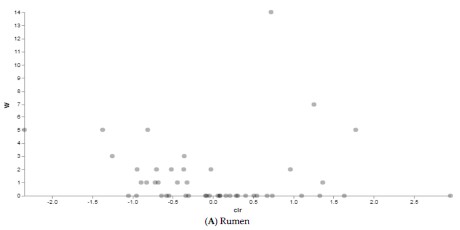
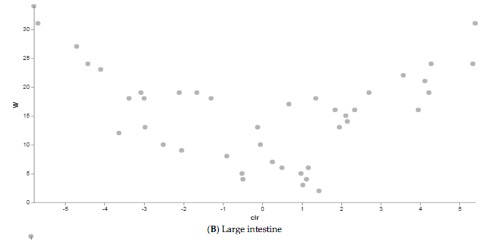
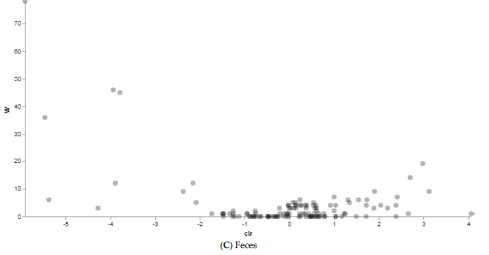
I risultati ottenuti sono in linea con quanto riscontrato attraverso le analisi precedenti, confermando una forte variabilità in termini di struttura del microbiota nell’intestino e nelle feci con specie che sono state identificate come differenzialmente abbondanti a seconda dell’alimentazione in questi siti corporei. Nel rumine, invece, anche le analisi ANCOM hanno confermato una minore eterogeneità fra i gruppi. Tale tendenza potrebbe essere spiegata dalle condizioni restrittive di questo sito corporeo. Quest’organo, infatti, funge da camera di fermentazione ed ha un pH compreso tra 5,5 e 6,9 ed una temperatura di 38-39 C° (2); per questo motivo solo alcune popolazioni microbiche riescono a sopravvivere.
In ultima analisi, la somministrazione della dieta alternativa, caratterizzata da un diverso rapporto CNS/PG, ha determinato un alterato il rapporto tra Firmicutes/Bacteroides a favore di questi ultimi, promuovendo, in particolare, lo sviluppo di Fibrocateres e Proteobacteria con ripercussioni sulla digeribilità della dieta. Quest’ultima è stata maggiore nelle bufale alimentate in maniera tradizionale. Tale fenomeno potrebbe essere dovuto alla presenza del licopene (molecole della classe dei polifenoli presente nel pomodoro) che reprime la presenza di Firmicutes abbassando l’efficienza di conversione degli alimenti (5,21). Tuttavia, recenti studi hanno dimostrato che la presenza di polifenoli presenti nei gusci della frutta secca favorisce le fermentazioni ruminali verso la produzione di propionato, andando a diminuire le emissioni di ammoniaca (NH3) e di metano (CH4) (22). Inoltre, a partire da questi risultati si potrebbe pensare di intervenire anche sul microbiota del GIT dei vitelli, cercando di stimolare la crescita di alcune specie a discapito di altre; questo potrebbe influire positivamente sulle problematiche legate ai disordini gastrointestinali neonatali.
In conclusione, i risultati ottenuti indicano che la dieta può influenzare fortemente il microbiota di diversi siti GIT e potrebbe quindi agire come fattore modulante per specifici microrganismi in grado di influenzare le prestazioni produttive degli animali adulti e non solo.
La presente nota è una sinossi del seguente articolo scientifico pubblicato sulla rivista Veterinary Science dove è riportata tutta la letteratura citata: Different Non-Structural Carbohydrates/Crude Proteins (NCS/CP) Ratios in Diet Shape the Gastrointestinal Microbiota of Water Buffalo. Rubina Paradiso, Giorgia Borriello, Sergio Bolletti Censi, Angela Salzano, Roberta Cimmino, Giorgio Galiero, Giovanna Fusco, Esterina De Carlo and Giuseppe Campanile. DOI: https://doi.org/10.3390/vetsci8060096.
Bibliografia
- Gomez DE, Galvão KN, Rodriguez-Lecompte JC, Costa MC. The cattle microbiota and the immune system: An evolving field. Veterinary Clinics: Food Animal Practice. 2019;35(3):485–505.
- Romagnoli EM, Kmit MCP, Chiaramonte JB, Rossmann M, Mendes R. Ecological aspects on rumen microbiome. In: Diversity and Benefits of Microorganisms from the Tropics. Springer; 2017. pag. 367–89.
- Bainbridge ML, Cersosimo LM, Wright A-DG, Kraft J. Rumen bacterial communities shift across a lactation in Holstein, Jersey and Holstein× Jersey dairy cows and correlate to rumen function, bacterial fatty acid composition and production parameters. FEMS Microbiology Ecology. 2016;92(5):fiw059.
- Pulido R, Muñoz R, Lemarie P, Wittwer F, Orellana P, Waghorn G. Impact of increasing grain feeding frequency on production of dairy cows grazing pasture. Livestock Science. 2009;125(2–3):109–14.
- Jami E, White BA, Mizrahi I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PloS one. 2014;9(1):e85423.
- Jewell KA, McCormick CA, Odt CL, Weimer PJ, Suen G. Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency. Applied and environmental microbiology. 2015;81(14):4697–710.
- Zou C, Gu Q, Zhou X, Xia Z, Muhammad WI, Tang Q, et al. Ruminal microbiota composition associated with ruminal fermentation parameters and milk yield in lactating buffalo in Guangxi, China—A preliminary study. Journal of animal physiology and animal nutrition. 2019;103(5):1374–9.
- Wallace RJ, Rooke JA, McKain N, Duthie C-A, Hyslop JJ, Ross DW, et al. The rumen microbial metagenome associated with high methane production in cattle. BMC genomics. 2015;16(1):1–14.
- Mizrahi I, Jami E. The compositional variation of the rumen microbiome and its effect on host performance and methane emission. Animal. 2018;12(s2):s220–32.
- Halmemies-Beauchet-Filleau A, Rinne M, Lamminen M, Mapato C, Ampapon T, Wanapat M, et al. Alternative and novel feeds for ruminants: nutritive value, product quality and environmental aspects. Animal. 2018;12(s2):s295–309.
- Bakshi M, Wadhwa M, Makkar HP. Waste to worth: vegetable wastes as animal feed. Cab Rev. 2016;11(012):1–26.
- Arco-Pérez A, Ramos-Morales E, Yáñez-Ruiz DR, Abecia L, Martín-García AI. Nutritive evaluation and milk quality of including of tomato or olive by-products silages with sunflower oil in the diet of dairy goats. Animal Feed Science and Technology. 2017;232:57–70.
- Krause D, Nagaraja T, Wright A, Callaway T. Board-invited review: rumen microbiology: leading the way in microbial ecology. Journal of animal science. 2013;91(1):331–41.
- Matthews C, Crispie F, Lewis E, Reid M, O’Toole PW, Cotter PD. The rumen microbiome: a crucial consideration when optimising milk and meat production and nitrogen utilisation efficiency. Gut microbes. 2019;10(2):115–32.
- Van Keulen J, Young B. Evaluation of acid-insoluble ash as a natural marker in ruminant digestibility studies. Journal of Animal Science. 1977;44(2):282–7.
- Prajapati VS, Purohit HJ, Raje DV, Parmar N, Patel AB, Jones OA, et al. The effect of a high-roughage diet on the metabolism of aromatic compounds by rumen microbes: a metagenomic study using Mehsani buffalo (Bubalus bubalis). Applied microbiology and biotechnology. 2016;100(3):1319–31.
- Catozzi C, Sanchez Bonastre A, Francino O, Lecchi C, De Carlo E, Vecchio D, et al. The microbiota of water buffalo milk during mastitis. PLoS One. 2017;12(9):e0184710.
- Leser TD, Amenuvor JZ, Jensen TK, Lindecrona RH, Boye M, Møller K. Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Applied and environmental microbiology. 2002;68(2):673–90.
- Huo W, Zhu W, Mao S. Impact of subacute ruminal acidosis on the diversity of liquid and solid-associated bacteria in the rumen of goats. World Journal of Microbiology and Biotechnology. 2014;30(2):669–80.
- Vasta V, Daghio M, Cappucci A, Buccioni A, Serra A, Viti C, et al. Invited review: Plant polyphenols and rumen microbiota responsible for fatty acid biohydrogenation, fiber digestion, and methane emission: Experimental evidence and methodological approaches. Journal of Dairy Science. 2019;102(5):3781–804.
- Cook GM, Rainey FA, Chen G, Stackebrandt E, Russell JB. Emendation of the description of Acidaminococcus fermentans, a trans-aconitate-and citrate-oxidizing bacterium. International Journal of Systematic and Evolutionary Microbiology. 1994;44(3):576–8.
- Sinisgalli C, Vezza T, Diez‐Echave P, Ostuni A, Faraone I, Hidalgo‐Garcia L, et al. The Beneficial Effects of Red Sun‐Dried Capsicum annuum L. Cv Senise Extract with Antioxidant Properties in Experimental Obesity are Associated with Modulation of the Intestinal Microbiota. Molecular Nutrition & Food Research. 2021;65(3):2000812.





Scrivi un commento
Devi accedere, per commentare.